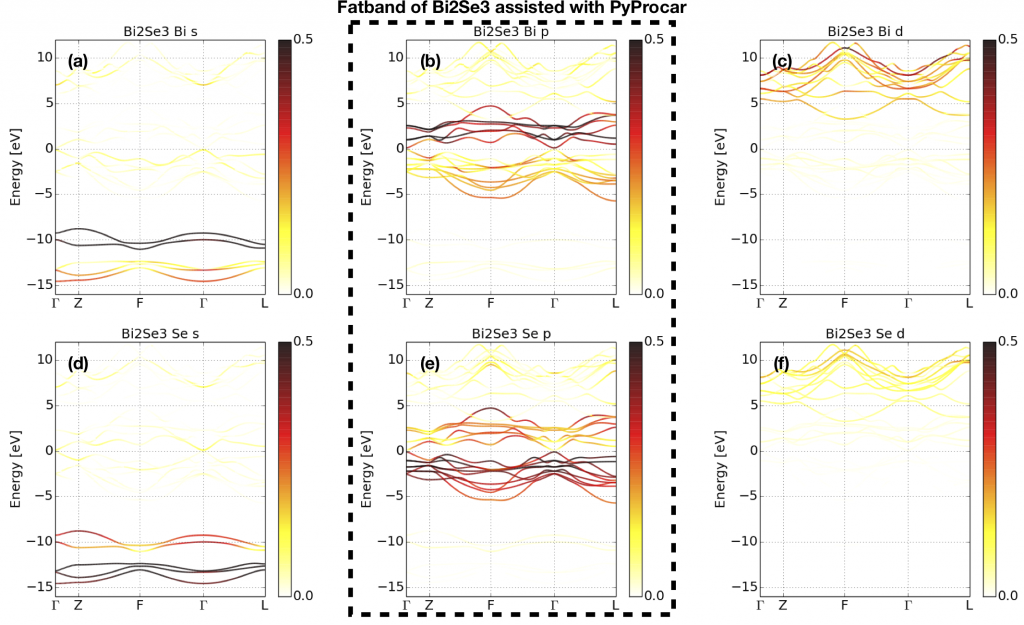
High-quality Wannier functions (WFs) are very useful for physical properties calculations after we obtained the self-consistent charge density from the first-principle calculations. However, for the beginners, it’s not easy to obtain good WFs. Here we would like to introduce a standard way to construct high-quality Wannier functions with first-principle software package and software Wannier90. Here we don’t focus on constructing maximum localized Wannier functions (MLWFs). We only want to construct a good tight-binding model based on WFs that are able to reproduce the band structure in the energy range we are interested in. Here are four criterions of high-quality WFs.
- 1. Perfect fitting to the DFT bands in the energy range you are interested in.
- 2. Keep the atomic orbital symmetry.
- 3. Well localized.
- 4. As small number of WFs as possible.
There are three important steps to reach four criterions.
- 1. Choose projectors.
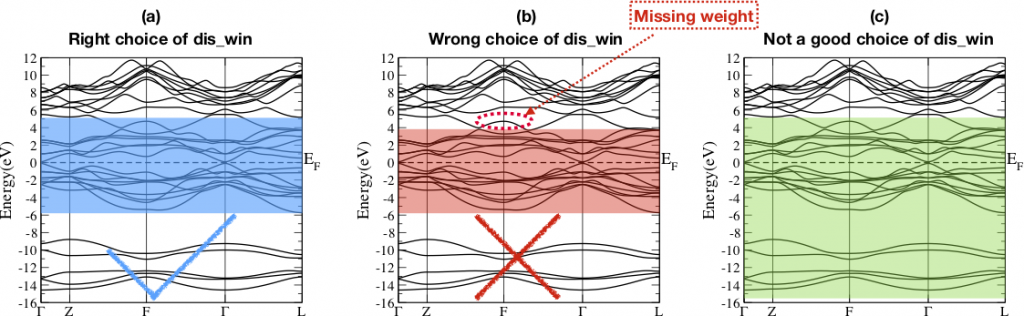
- 2. Choose disentanglement energy window.
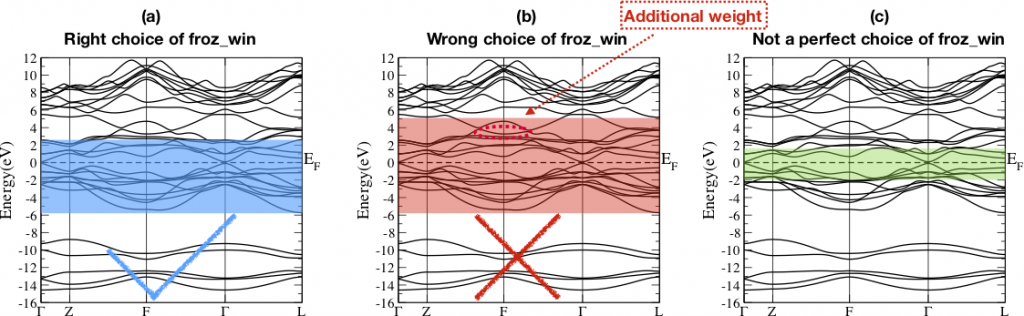
- 3. Choose frozen energy window.
I would like to take Bi2Se3 as an example. In order to save the computation cost, we don’t take into account the spin-orbit coupling (SOC) effect.
1. Choose projectors.
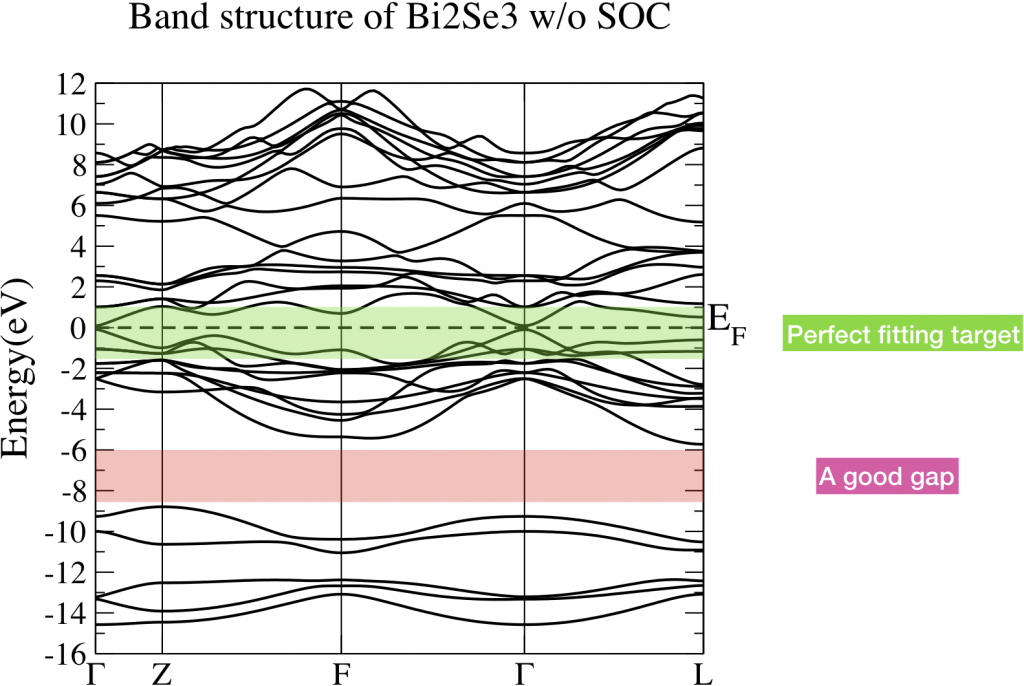
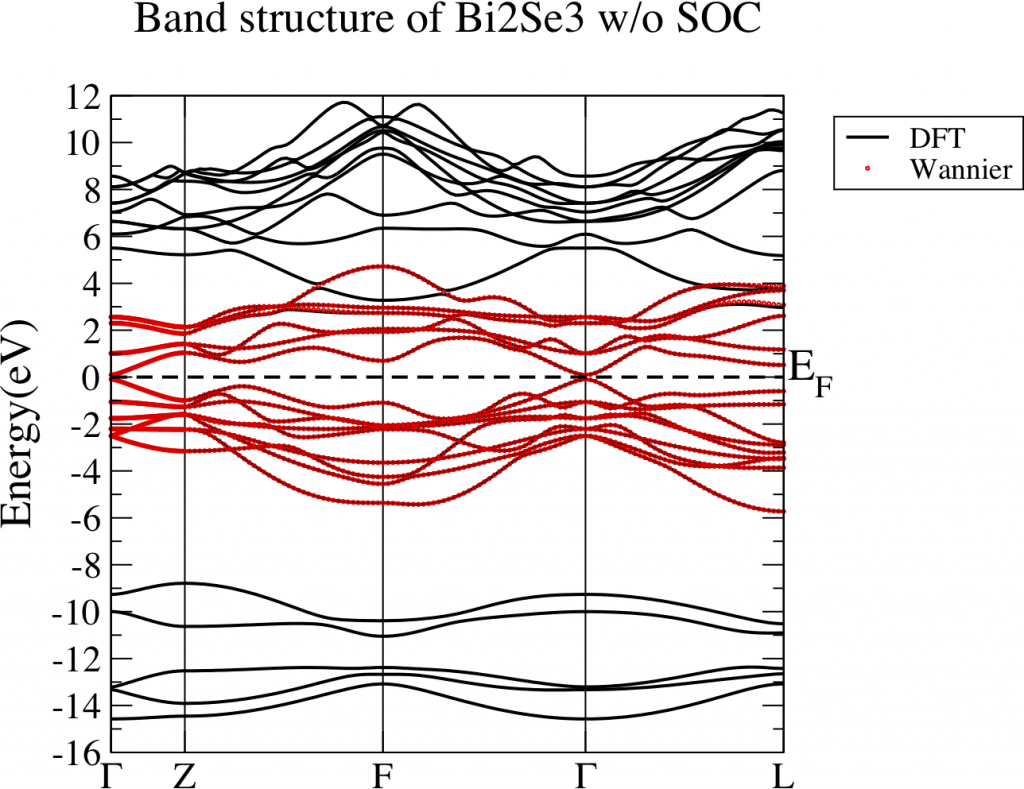
Let’s perform energy band analysis first. Fig 1 is the band structure of Bi2Se3 without considering SOC. The green shaded part is the energy range which we care about. We want to construct a tight binding model which can well reproduce the energy bands in that interested range. However, those band is connected with other bands below and above. Noticeably, there is a big gap around -8.4eV and -6eV. This is a good sign since the bands below this gap is not hybridized with the bands we are interested in. In this case, those bands below the gap are not necessary to be presented in our tight binding model. Eventually, the number of WFs reduce. It fulfills the fourth criterion.